抗生素抗藥性危機的關鍵挑戰之一是尋找新的抗菌化合物。雖然過去80年多數抗生素來自真菌和細菌(尤其是放線菌)所產生的天然產物,但由於常重複發現已知藥物骨架,這些來源逐漸不被看好。目前普遍認為放線菌已被過度開採,難以發現新穎化合物。本研究展示利用改良的分餾技術,能富集先前被忽略的少量產物,即使是研究已久的抗生素生產菌株,也能提供具有獨特作用機制的新化學骨架。
透過分餾土壤細菌天然產物萃取物庫,我們發現著名抗生素氧四環素的來源菌Streptomyces rimosus能產生一種環狀酯肽抗生素,命名為manikomycin。manikomycin能殺死多重抗藥性腸道菌科細菌,且不受臨床常用抗生素抗藥機制影響。生化、遺傳及結構分析顯示,manikomycin結合細菌核糖體大亞基的E位點,阻止tRNA 3′端進入E位點,有效抑制蛋白質合成轉位步驟,且具序列上下文特異性。據我們所知,manikomycin是首個針對細菌核糖體大亞基E位點的抗菌劑,凸顯其作為新抗生素開發先導化合物的價值。
過去80年,微生物天然產物,特別是放線菌衍生物,一直是抗菌劑的主要來源。這些抗生素是透過測試生產菌粗萃取物抑制細菌生長的能力發現,稱為Waksman平台。儘管該方法初期成效顯著,但隨著時間推移,因常重複發現常見化學骨架,加上目標導向高通量篩選合成化合物的興起,其效用逐漸降低。然而,多重抗藥病原體的崛起及目標導向篩選的有限成功,重新激起對微生物天然產物作為新抗菌劑來源的興趣。放線菌基因組中未開發的潛力促使對這些微生物的新藥先導物的研究熱潮。
Streptomyces基因組中富含潛在編碼抗菌化合物的生物合成基因簇(BGC),但因表達量低、產物多樣且分析方法不足,僅少部分化合物被分離。改良分餾天然產物萃取物能分離活性重疊的成分,如兩種不相關抗生素,成為挖掘隱藏抗生素的策略。本研究應用此策略,發現一種新型環狀酯肽抗生素manikomycin(MKM),源自1950年已知產氧四環素的Streptomyces rimosus。MKM展現獨特作用機制:結合細菌核糖體大亞基E位點,干擾轉位,且對抗藥性革蘭氏陰性病原體有效,且不受臨床菌株抗藥機制影響,提供新抗生素化學骨架。
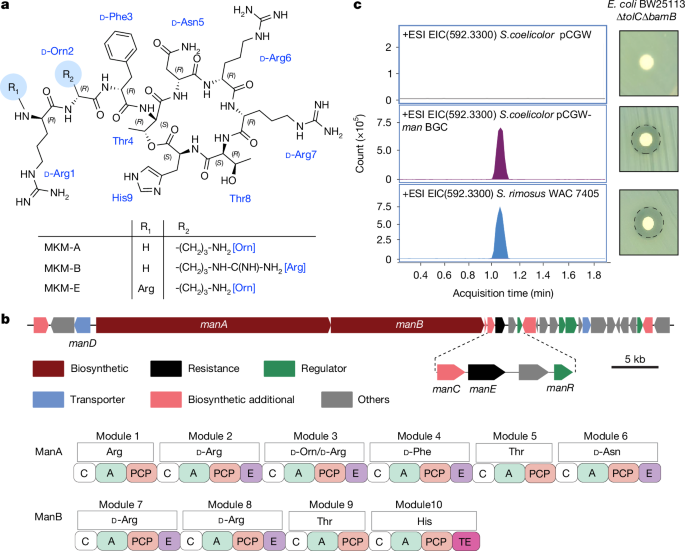
我們篩選了255株細菌菌株的甲醇萃取物,主要為多樣放線菌,來自土壤樣本。萃取物先按極性分餾,提高發現活性成分機率。活性分餾物經代謝組學指導去重,避免重複發現已知天然產物。以對革蘭氏陰性抗生素敏感的大腸桿菌BW25113 ΔtolC ΔbamB篩選。Streptomyces rimosus WAC 7405萃取物經尺寸排阻色譜分餾,獲得不同抗菌活性分餾。質譜分子網絡分析識別出氧四環素及其衍生物於部分分餾中,但活性分餾1和2未匹配已知化合物。進一步生物活性導向純化鑑定出MKM家族,為新型陽離子環狀酯肽。結構搜尋確認MKM骨架在SciFinder及PubChem中均無相似分子。
MKM名稱源自印地語及旁遮普語「manik」,意指珍貴寶石,象徵其稀有及獨特作用。最豐富為九肽MKM-A。利用質譜及一維/二維核磁共振光譜確定結構,氨基酸立體化學由Marfey分析驗證。其他較少見變體包括九肽MKM-B、八肽MKM-C及MKM-D及十肽MKM-E。MKM為酯肽,透過C端組氨酸與第4位蘇氨酸側鏈羥基形成環狀酯鍵。MKM-A、MKM-D及MKM-E在第2位含d-鳥氨酸,MKM-B及MKM-C則為d-精氨酸。
基因組測序及antiSMASH分析鑑定出man BGC,推測為MKM生物合成基因簇,包含兩個非核糖體肽合成酶ManA及ManB,分別含6及4個模組,合成10個氨基酸肽鏈,對應MKM-E結構。最後步驟為C端組氨酸與Thr4環化釋放,可能由末端硫酯酶催化。模組中表現的表異構酶域預測與實驗立體化學吻合。
將67 kb MKM BGC轉入異源宿主Streptomyces coelicolor M1154,成功分泌MKM,質譜及抗菌活性驗證基因簇功能。後續研究主要使用MKM-A。
MKM帶正電,可能具膜破壞活性,但實驗顯示不破壞細菌膜,也無細胞形態改變,故推測作用於細胞內目標。大腸桿菌BW25113在亞抑制濃度MKM存在下傳代,選出高抗性突變株,最低抑菌濃度(MIC)提高16至32倍。抗性頻率極低。全基因組測序發現抗性株中sbmA基因失能,該基因編碼膜內肽轉運蛋白,參與其他肽抗生素攝取。刪除sbmA基因使MIC增加4倍。刪除另一肽轉運蛋白YejABEF組件亦使MIC增加2至8倍,顯示MKM透過多條路徑進入細胞,作用於細胞內目標。
另一抗性株基因組測序發現rpmI基因(編碼大核糖體亞基蛋白bL35)出現無義突變,指向核糖體為MKM作用目標。大多數核糖體抗生素作用於rRNA,但因rRNA基因冗餘,rRNA抗性突變罕見。為驗證MKM抑制核糖體,我們在單拷貝rRNA操作子且過表達sbmA的抗生素超敏感株SQ110ΔtolC pZ-sbmA中選出抗性株。數百抗性菌株出現,頻率約10^-7。11株隨機抗性株23S rRNA基因測序顯示10株在4連續腺嘌呤區域缺失一腺嘌呤,1株缺失一胞嘧啶,MIC均提升16倍以上。這些突變及bL35蛋白均位於細菌大核糖體亞基E位點,該位點先前未被發現為抗菌劑結合處。
利用大腸桿菌無細胞轉譯系統證實MKM強效抑制細菌蛋白質合成(IC50=0.6 µM)。相比之下,MKM對兔網狀紅細胞裂解液轉譯抑制較弱(IC50=9.2 µM),約為細菌系統15倍,且遠高於已知真核E位點抑制劑環己酰亞胺。顯示MKM為選擇性抗菌蛋白質合成抑制劑。
高解析度結構分析顯示MKM結合於轉譯中核糖體。利用toeprinting技術定位MKM使核糖體停滯於ermBL mRNA第三密碼子。冷凍電子顯微鏡分析顯示主要核糖體狀態含A及P位tRNA,無E位tRNA,解析度達2.4 Å。兩處額外密度明確為MKM。主要結合位於50S亞基E位點,另一位點遠離功能中心。MKM環狀肽核心插入23S rRNA H13、H21及H88螺旋形成的口袋,帶電殘基與rRNA糖磷酸骨架形成多重氫鍵及水介導相互作用。抗性突變位於結合位點附近,推測透過局部構象改變破壞MKM結合。bL35蛋白不直接接觸MKM,但與rRNA互動,bL35突變可能透過變構效應影響MKM結合。
MKM在E位點的結合阻擋去酰化tRNA CCA端進入,特別是Phe3與E位tRNA末端腺嘌呤重疊。真核及古菌核糖體E位點為多種翻譯抑制劑目標,但不抑制細菌翻譯。真核特有蛋白eL42阻擋MKM結合,解釋MKM對人類細胞低毒性及弱抑制真核翻譯。
MKM阻礙核糖體形成混合狀態,阻止P位tRNA轉位至E位,進而阻止A位tRNA轉位至P位。冷凍電鏡顯示MKM使核糖體停留於轉位前狀態。體外轉位實驗證實MKM抑制轉位,類似已知轉位抑制劑negamycin。
toeprinting分析顯示MKM在ermBL mRNA第三密碼子使核糖體停滯,但亦在其他特定mRNA位置造成停滯,顯示其抑制作用受mRNA序列上下文影響。細菌細胞核糖體剖析顯示MKM處理細胞早期密碼子處核糖體積聚,常見於延伸抑制抗生素。停滯位置富含脯氨酸密碼子,脯氨酸蛋白質合成困難,可能使E位tRNA較易解離,利於MKM結合。偏好位點的肽鏈末端富含異亮氨酸、亮氨酸及脯氨酸,顯示肽鏈依賴性核糖體停滯。相反,蘇氨酸密碼子在停滯位點及前方明顯減少,實驗插入三個蘇氨酸密碼子mRNA顯示MKM抑制較弱,推測tRNA Thr結構或轉位特性影響MKM作用。
抗生素生產菌需避免自我毒性,常透過rRNA甲基化產生免疫。本研究發現MKM BGC中含putative rRNA甲基轉移酶基因manE,僅存在於含MKM BGC的S. rimosus菌株。表達manE於大腸桿菌使其對MKM抗性提升32倍以上,對其他翻譯抑制劑無影響,顯示ManE修飾MKM作用位點rRNA。引物延伸分析及液相色譜質譜確認ManE在23S rRNA C2395位點2′-O-甲基化,該位點與MKM結合直接相關,甲基化阻斷MKM氫鍵形成,解釋抗性機制。
MKM對革蘭氏陰性腸道菌科(大腸桿菌及肺炎克雷伯菌)及分枝桿菌有效,對其他革蘭氏陰性及大多數革蘭氏陽性菌無效,推測因細胞攝取限制。MKM結合位點在多種細菌核糖體保守,故抗菌活性缺失非因靶點差異。MKM不受臨床菌株常見翻譯抑制抗性機制影響。
MKM無溶血性及對HEK293、HepG2細胞無明顯毒性。人血外感染模型中,MKM使肺炎克雷伯菌負荷降低約1000倍。小鼠感染模型初步無效,可能因藥代動力學問題。秀麗隱桿線蟲感染模型中,MKM顯著提升感染存活率,效果與多黏菌素B相當。
S. rimosus同時產生多種抗菌化合物,較低表達的抗生素活性可能被主導抗生素掩蓋。透過細緻分餾,首次發現MKM,顯示許多未知抗生素仍被已知抗生素掩蓋。
MKM為新抗生素化學骨架。ManAB NRPS模組預測產生富含精氨酸的十肽MKM-E。模組3 Stachelhaus基序突變導致Orn與Arg可變置換,顯示腺苷酸化域底物多樣性。
MKM為首個針對細菌核糖體大亞基E位點的抗生素。E位點在翻譯中重要性長期爭論,MKM提供研究該位點功能工具。MKM結合模式適用於其他MKM變體,推測機制相同,具序列上下文特異性。
雖有多種真核及古菌E位點抑制劑,無一抑制細菌翻譯,因E位點結構差異。此差異解釋MKM對哺乳動物低毒性及選擇性。且MKM為首個細菌大亞基E位點抑制劑,臨床菌株常見抗性機制無法抵抗MKM。
真核E位點抑制劑作用機制多樣,環己酰亞胺阻斷延伸核糖體,乳酸咪唑霉素則捕捉起始核糖體。MKM表面類似環己酰亞胺,阻斷延伸核糖體,但作用機制不同。環己酰亞胺體積小,可與E位tRNA共存,MKM體積大,幾乎完全阻塞E位,阻止去酰化tRNA進入及P/E混合狀態形成,阻斷轉位。部分P/E混合狀態可能仍形成,如tRNA Thr,解釋其較不易被MKM抑制。
大多數延伸抑制劑抑制強度受mRNA序列及新生肽鏈影響。MKM不直接與mRNA或肽鏈接觸,且可能僅與E位tRNA CCA端接觸,故其序列特異性令人意外。推測與翻譯動力學或E位tRNA停留時間相關。快速E位tRNA解離、A位tRNA慢速進入或肽鍵形成緩慢,均利於MKM結合。反之,E位tRNA長時間停留或特定tRNA競爭,降低MKM停滯機率。蘇氨酸密碼子低停滯率可能因tRNA Thr結構特性。未來研究可探討此機制。
為預防抗藥性,了解細菌如何避免MKM抑制至關重要。結果顯示肽轉運蛋白突變提供低度抗性。改變MKM化學性質,調整電荷,並使其細胞攝取不依賴肽轉運蛋白,或可克服抗性並擴大抗菌範圍。MKM生產菌透過ManE甲基轉移酶2′-O-甲基化23S rRNA C2395位點避免自毒。若MKM成藥,manE基因可能被病原菌獲得,但高解析結構提供改造MKM(如His9位點)以避開ManE抗性的路徑。
MKM-A在小鼠急性耐受性良好,最高劑量220 mg/kg/日,但感染模型無效,藥代動力學評估顯示血漿穩定性佳,卻血中濃度低且清除快,半衰期約36分鐘。顯示缺乏療效非因藥理活性不足,而是暴露不足。後續將優化藥物性質。MKM多陽離子特性可能帶來腎毒性及藥代限制,需謹慎處理,但骨架易於化學修飾,具調整潛力。相關類似物開發中。
MKM提供新抗生素骨架,具新穎核糖體作用位點及對革蘭氏陰性病原體活性,具開發潛力。本研究證明重新探索「老」抗生素生產菌,微生物基因組中仍蘊藏豐富抗生素化學多樣性。
本研究使用菌株及質體詳見補充資料。天然產物萃取物庫以384孔板篩選,使用高通量液體處理系統,37°C培養20小時,OD600測量生長。S. rimosus WAC 7405常規培養於TSB培養基,後接種至ASM培養基培養4天。活性化合物由條件培養基與Diaion HP-20樹脂混合吸附,過濾後用甲醇萃取,旋轉蒸發乾燥,重溶於水後以Sephadex LH-20分離,活性分餾再以液相色譜-質譜分析及GNPS分子網絡鑑定。活性分餾1及2進一步反相色譜純化,分離出MKM-A及MKM-B。